
Research
Single-Molecule Studies of DNA Damage Tolerance and Repair
Our laboratory studies the molecular mechanisms by which cells duplicate, repair and organize chromosomes. Traditional ensemble biochemistry has provided remarkable insight into the various activities of individual proteins and their collective action in these processes. However, probing the dynamics of protein-protein interactions is extremely difficult in bulk experiments as the stochastic appearance and disappearance of transient intermediates tends to obscure any observable when averaged over the ensemble. Single-molecule methods are a powerful way to overcome this problem by observing the individual trajectories of proteins as they function. Major areas of current research include:

DNA double strand breaks (DSBs) are extremely toxic lesions that can arise spontaneously or can be induced by agents such as ionizing radiation or endonucleases involved in programmed genome rearrangements. For the majority of the cell cycle DSBs are repaired by NHEJ, a process that robustly ligates even damaged or incompatible DNA ends, albeit in a way that can generate insertion or deletion mutations. We are using single-molecule FRET approaches to directly visualize the repair of DSBs in reconstituted systems and in vertebrate cell free extracts. We have demonstrated that end synapsis passes through at least two structurally distinct states and have identified the core NHEJ factors required to form these states. In current work, we are elucidating the network of intermolecular interactions that holds these states together and how error-prone end processing enzymes are regulated during repair.
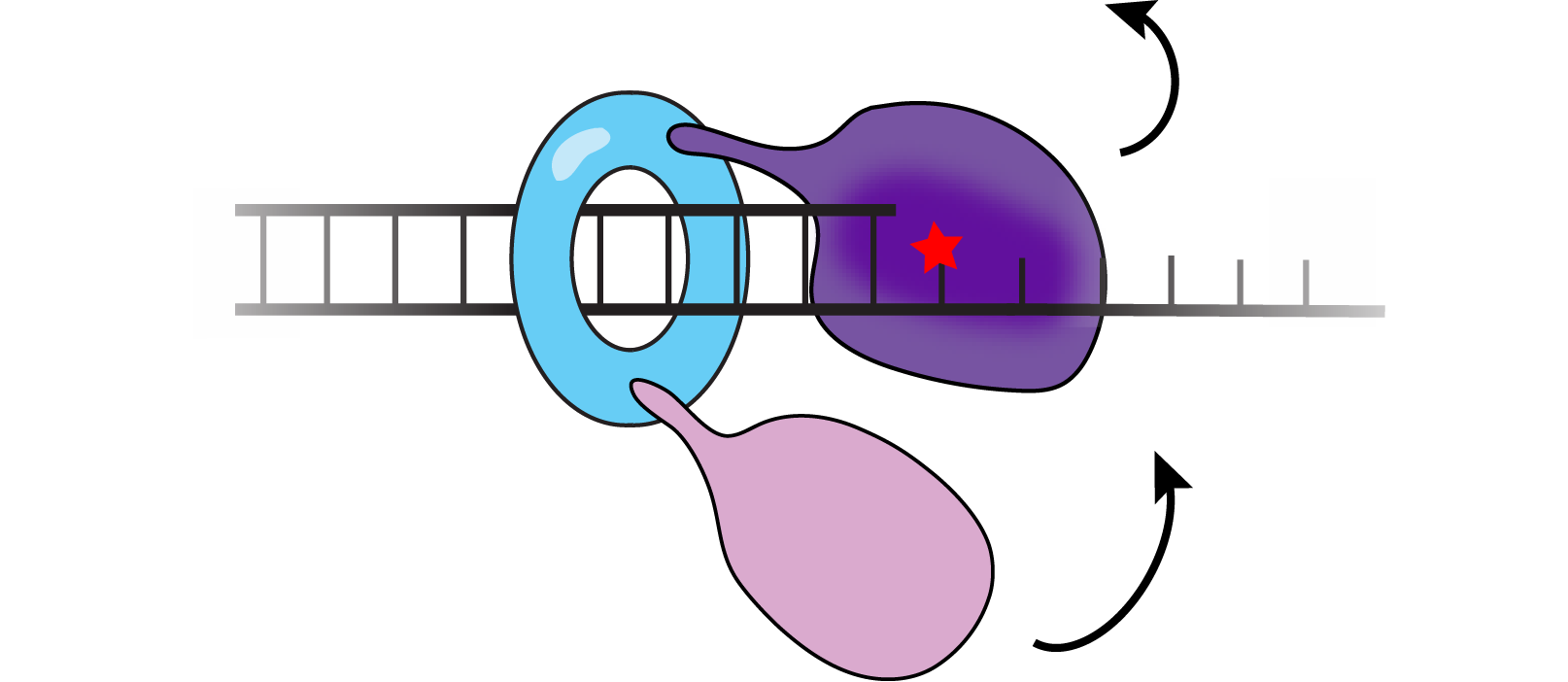
We are interested in how cells regulate the access of low-fidelity polymerases to the replication fork as their misuse leads to genome instability. In translesion synthesis (TLS), error-prone TLS polymerases are recruited to sites of DNA damage to carry out strand extension over DNA lesions that block the progress of the replisome. Using E. coli as a model, we have established a comprehensive picture of how error-prone translesion polymerases are regulated in bacteria. Most notably, our work describes how translesion polymerases are normally excluded from the replication machinery during DNA synthesis yet rapidly recruited to the replisome upon DNA damage. We have characterized key intermediates in the process of polymerase exchange and have identified distinct intermolecular interactions that act to either suppress TLS or enable it. Our work has identified novel intermolecular interactions which impact mutagenesis in bacteria.
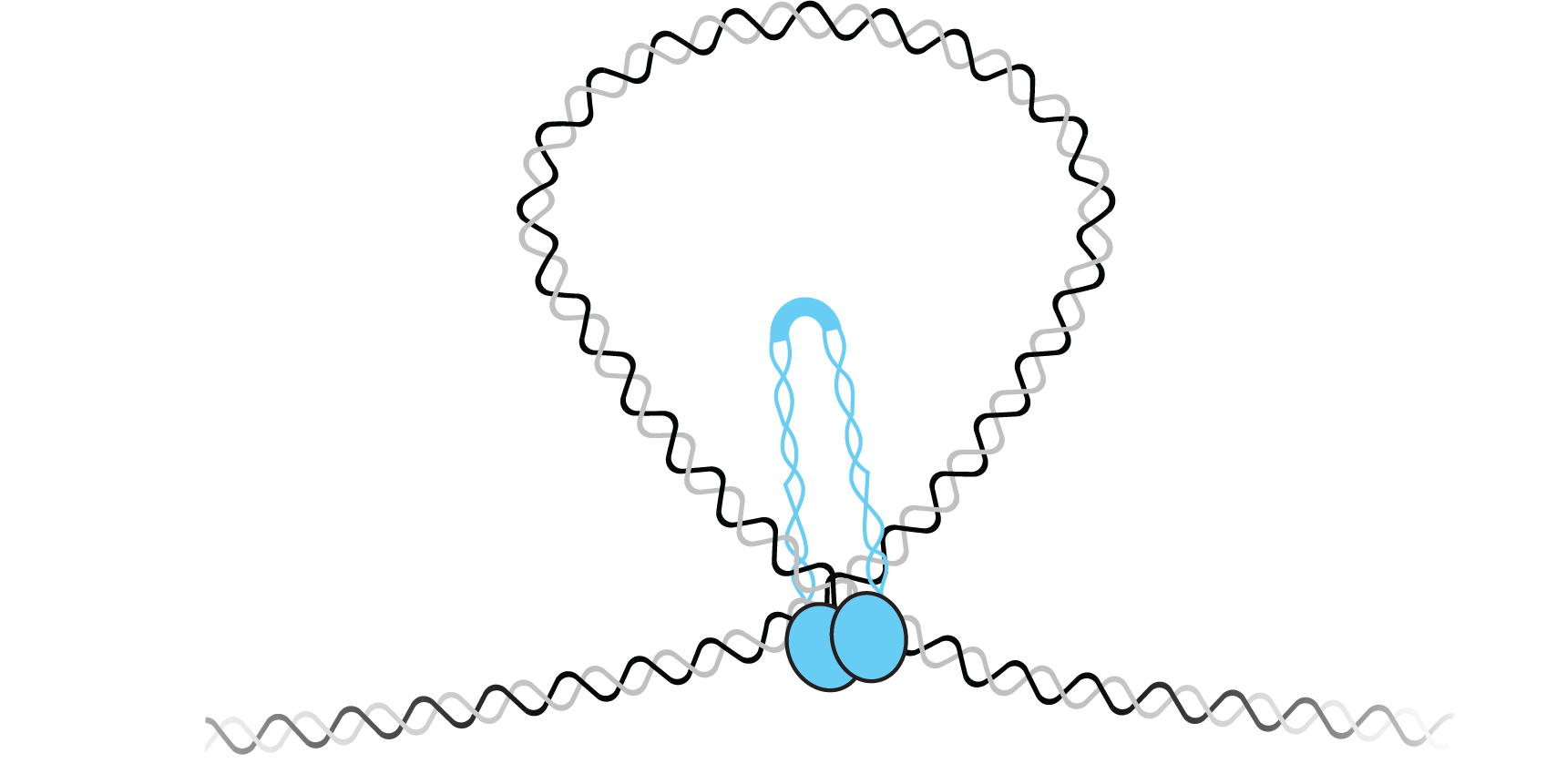
In the field of genome organization, our laboratory has advanced the understanding of how DNA binding proteins contribute to chromosome compaction and segregation. We have developed new approaches to observe the conformation of individual DNAs and to watch protein-DNA binding in the absence of protein labeling. We have applied these approaches to study a diverse set of DNA binding proteins in both bacterial and eukaryotic systems. In bacteria, we showed that the highly conserved ParB uses DNA bridging interactions to facilitate chromosome segregation. Furthermore, we showed that the bacterial SMC forms higher order structures that condense DNA. Working with the laboratory of Steve Jacobsen, we showed that MORC-1 - a member of the highly conserved GHKL type ATPases involved in gene silencing - compacts DNA through a topological entrapment mechanism.
Outside of our genome maintenance efforts, we collaborate with members of the Harvard Medical School community to better understand the structural dynamics of membrane proteins. With the laboratory of Dr. Stephen Blacklow, we showed that mechanical force is the physiological signal that initiates Notch signaling. We have also worked with Dr. Tom Rapoport to use single-molecule FRET assays to understand how ATP hydrolysis within the SecA ATPase is coupled to polypeptide translocation. Ongoing efforts with the laboratory of Dr. Andrew Kruse are focused on the structural dynamics of the SEDS-family peptidoglycan polymerase – a critical factor in the assembly of the bacterial cell wall and an important target in next-generation antibiotic development.